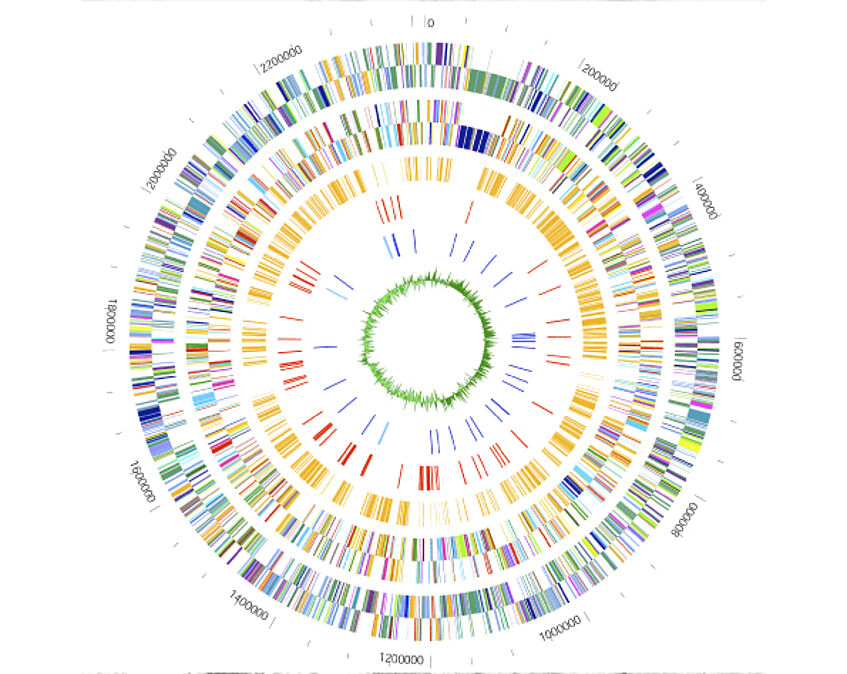
Comparative genomics
Life on earth has evolved for some billion years. Living organisms are found in virtually every environment, surviving and thriving in extremes of heat, cold, radiation, pressure, salt, acidity, and darkness.
Often in these environments, only “simple” microorganisms are found and the only nutrients come from inorganic matter. The diversity and range of environmental adaptations indicate that even tiny microbes long ago "solved" many problems for which scientists and engineers are still actively seeking solutions.
These secrets are enclosed in the genomes, which encode the construction plans of cells and organisms. The first complete bacterial genome was deciphered in 1995. Since then the number of complete genomes sequenced is exponentially growing. Only powerful computers and sophisticated bioinformatics software allows us to investigate these massive data. At CUBE we are involved in many genome-sequencing projects. We also create new software for the annotation and analysis of genome sequences.
The accessibility of almost complete genome sequences of uncultivable microbial species from metagenomes necessitates computational methods predicting microbial phenotypes solely based on genomic data. We therefore investigate how comparative genomics can be utilized for the prediction of microbial phenotypes. The PICA framework facilitates application and comparison of different machine learning techniques for phenotypic trait prediction. We have recently improved and extended PICA's support vector machine plug-in and suggest its applicability to large-scale genome databases and incomplete genome sequences. We have demonstrated the stability of the predictive power for phenotypic traits, not perturbed by the rapid growth of genome databases.Most of the traits can be reliably predicted in only 60-70% complete genomes. These results suggest that the extended PICA framework, as available online at phendb.org, can be used to automatically annotate phenotypes in near-complete microbial genome sequences, as generated in large numbers in current metagenomics studies. We further extend and to improve this approach in order to predict as many traits as possible.